Clustering viral sequences based on shared proteins
Overview
Teaching: 0 min
Exercises: 60 minQuestions
Can we cluster viral sequences based on shared protein clusters?
Objectives
Identify putative crAssphage bins
Another way to classify your genomes is to cluster them based on the gene content of the genomes. We will first visualize the protein content of the viral bins using a sort of heatmap that reflects the phylogenetic profiles of the different protein clusters generated on day 3. For this we are going to use the MMSeqs2 protein clusters (PCs) you made in the previous day from your binned sequences.
First, open a terminal and make a new folder for today’s results to keep things organised:
$ cd /path/of/viromics/folder/
$ mkdir day4
$ cd day4
We provide an R script for this part of the analysis. However, if you are familiar with making heatmaps and clustering data in R (or any other programming language), please feel free to read along and make your own script.
# Download the script from github:
$ wget https://raw.githubusercontent.com/MGXlab/Viromics-Workshop-MGX/gh-pages/code/day4/clustering.R
Tidyverse should have installed by now: go back to Rstudio and check. If you have trouble installing Tidyverse please contact any of the assistants.
Create a new project in Rstudio (file > create new project > Existing directory) and select the
/path/of/viromics/folder/day4 folder.
Open the clustering.R script you just downloaded. It should be in the lower
right pane under “Files”
Question 1: Data exploration.
Follow the R script up until line 18. Make sure to change the file paths so that they point correctly, and read the comments to understand what you’re doing.
Take a moment to look at what was in the data you just loaded.
- What’s in each of the different columns?
- What information do you still lack to be able to cluster genomes based on protein content?
The RefSeq proteins have unique protein IDs (e.g. YP_009124822.1), but no identifier that tells us to which genome they belong.
We will download this information and some additional taxonomic metadata from
NCBI virus.
Go to Find Data > Bacteriophages (A):
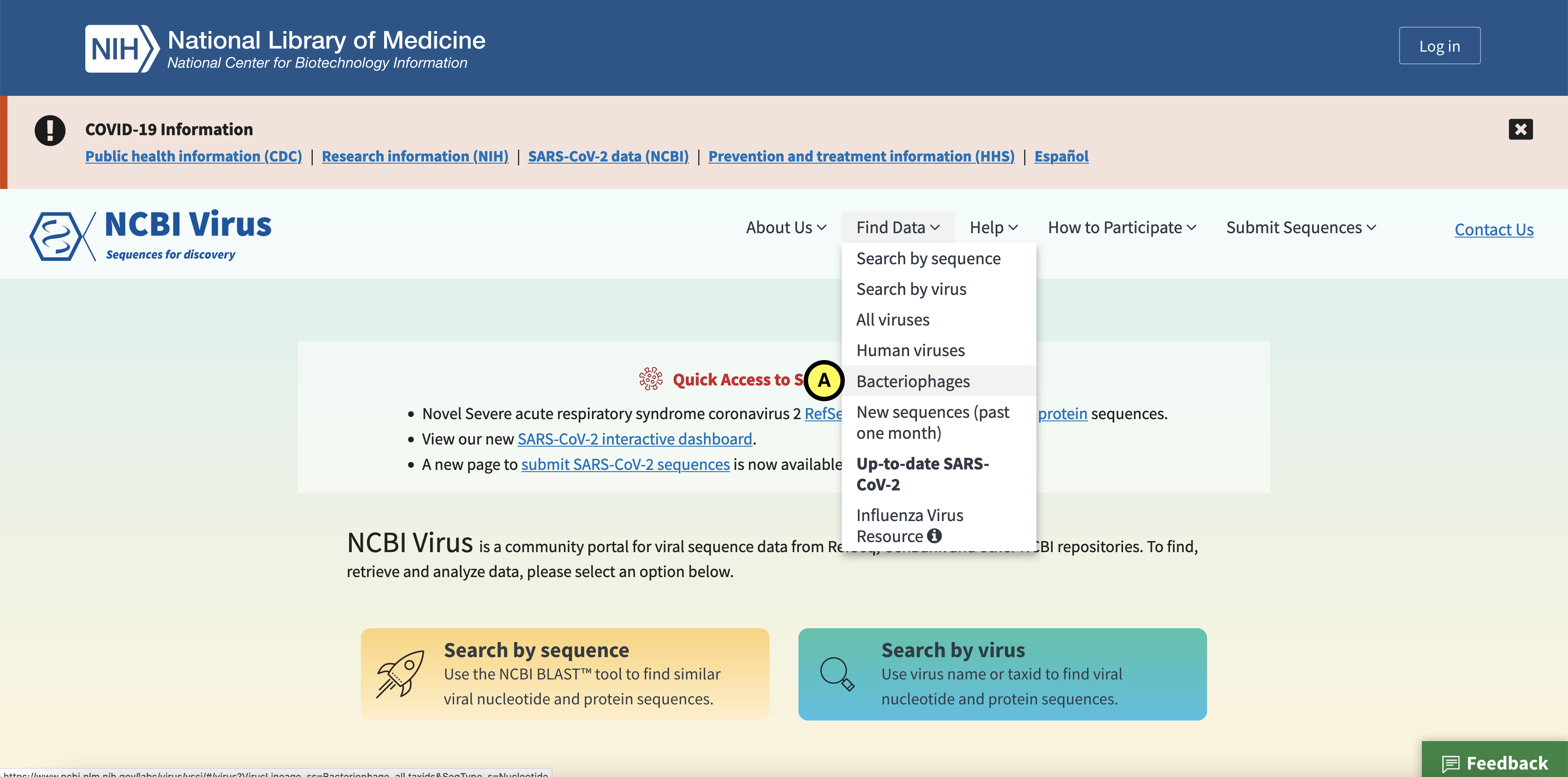
Click the Protein tab (B), and in the left (C) select complete and partial RefSeq genome completeness. Next click “Download” (D):
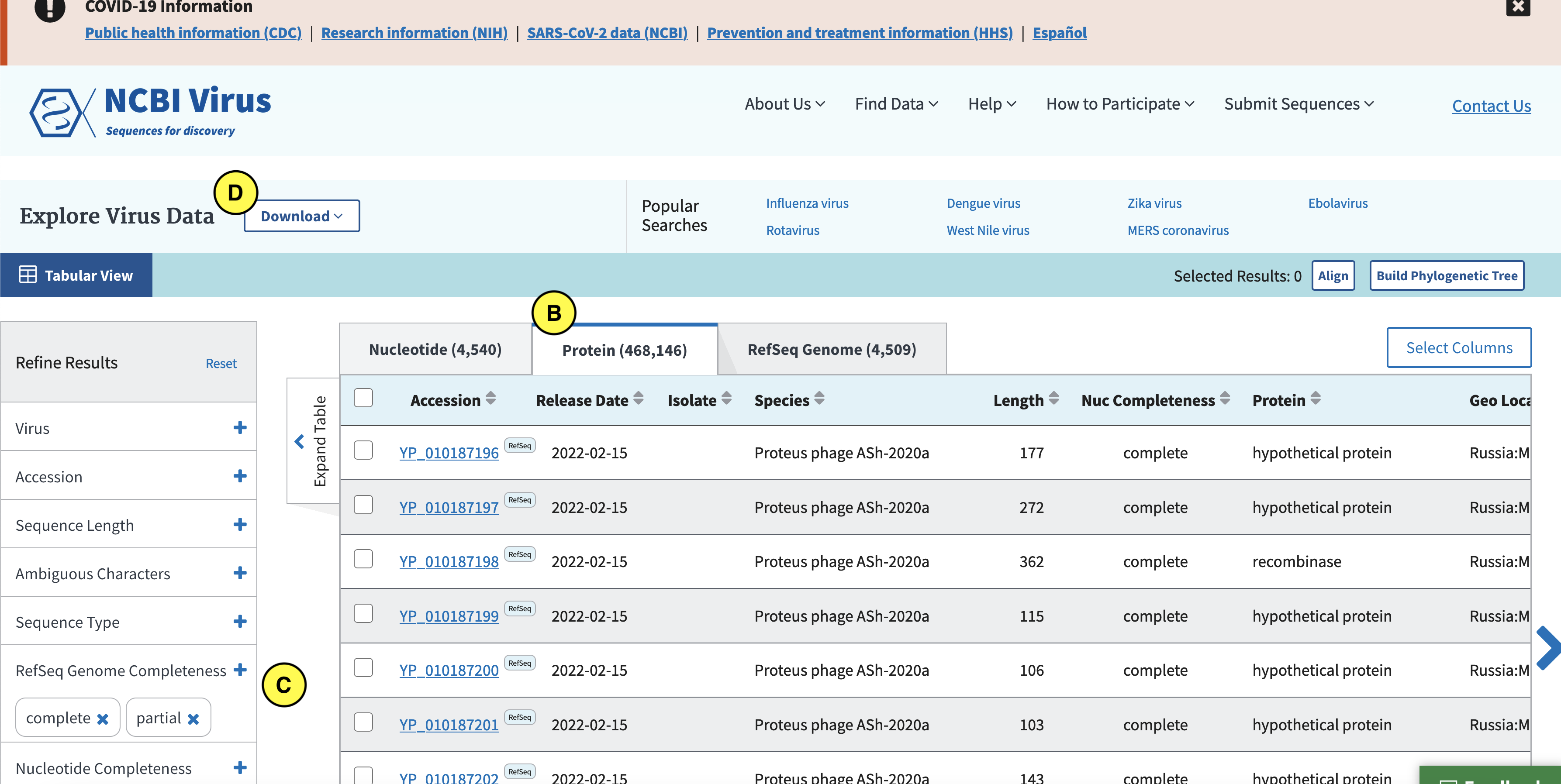
In the screen that pops up click Current table view results, csv format (E):
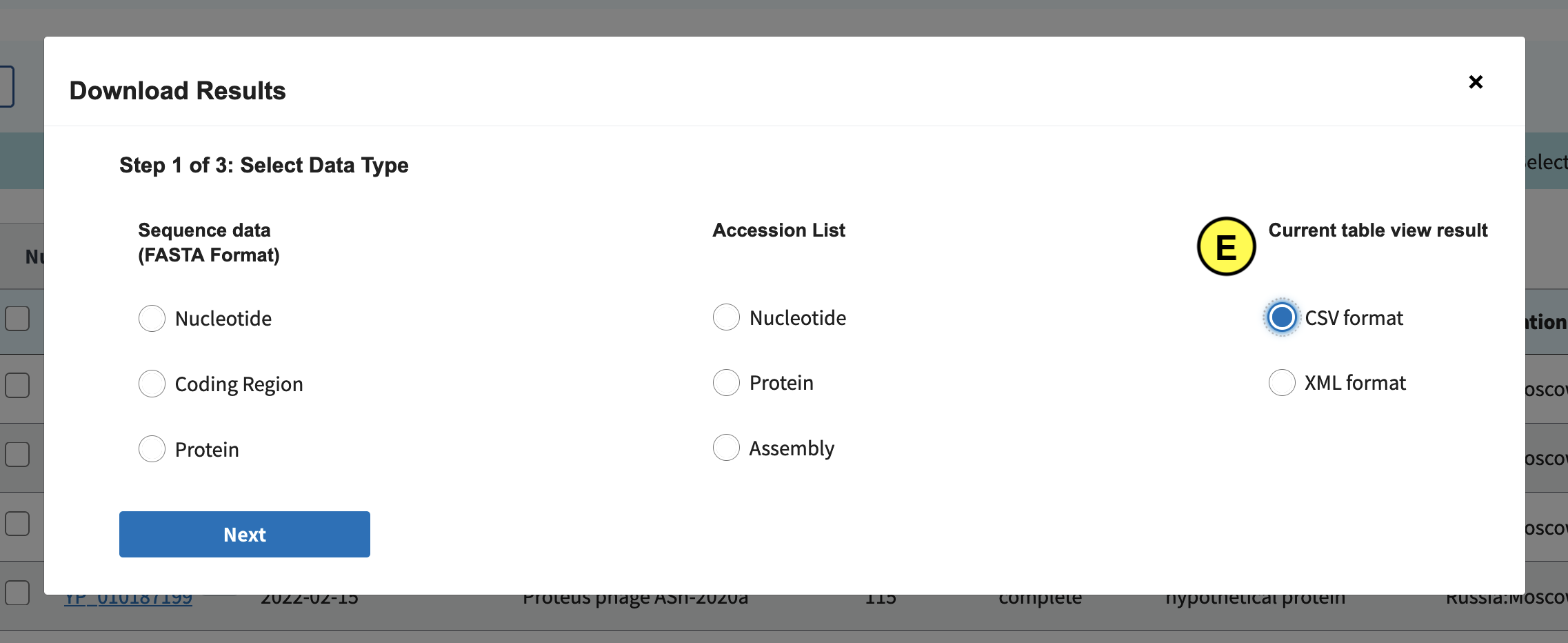
Download all records:
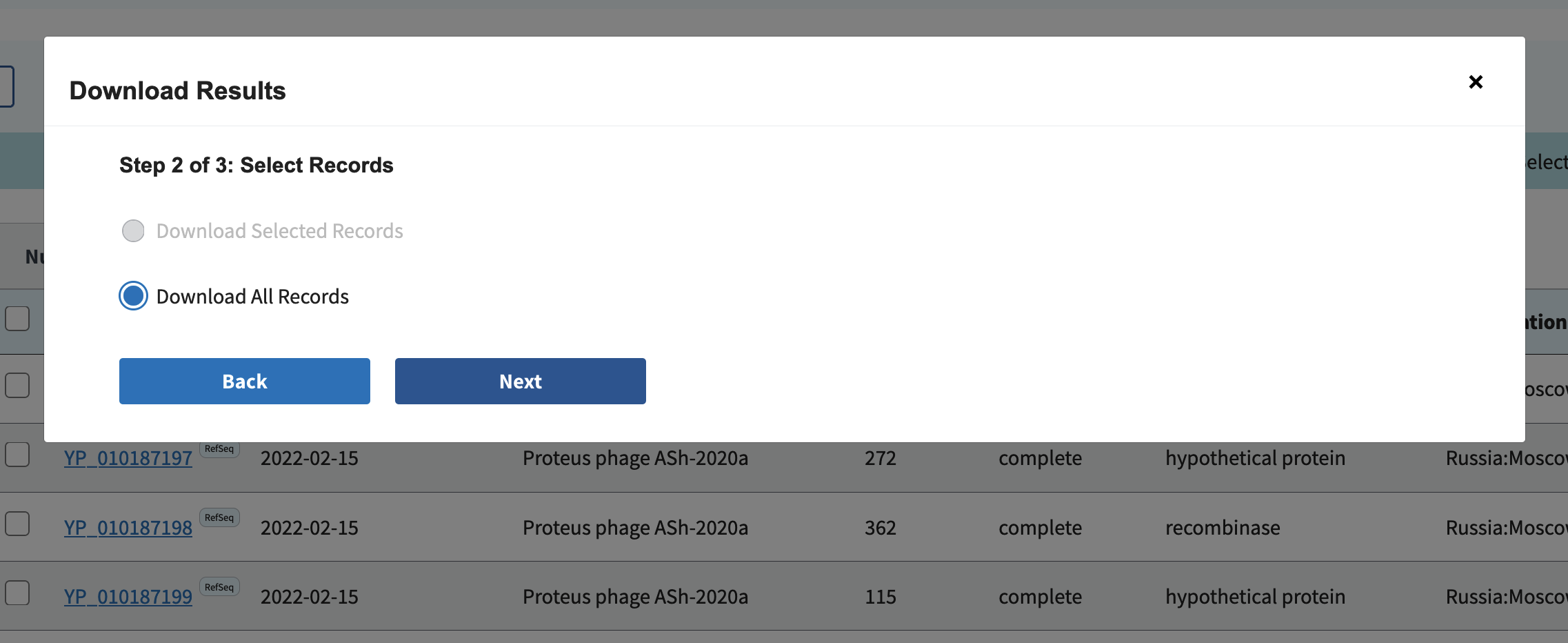
Select Accession, Species, Genus, Family, Length, Protein, and Accession with version (F):
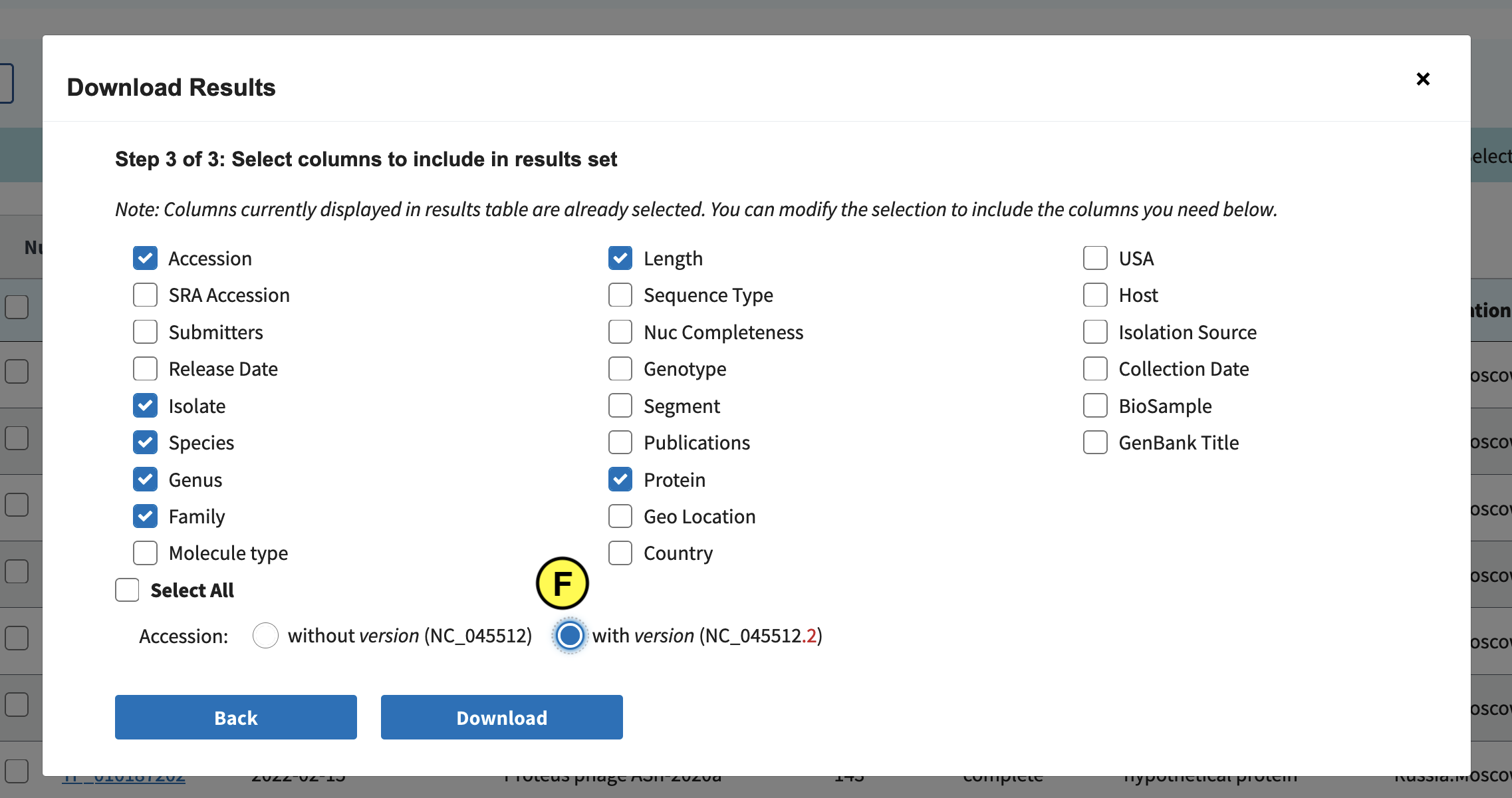
Finally click Download. Downloading might take long, so as a backup we’ve also included the file in the Github repo:
# Download the metadata from github:
$ wget https://raw.githubusercontent.com/MGXlab/Viromics-Workshop-MGX/gh-pages/data/sequences_bac.csv
Question 2: Clustering contigs based on shared protein clusters
Go back to the R script and follow the steps up to line 87.
Look at the heatmap. Are there any core viral genes? Can you find closely related bins?Solution
We observe:
- sparse clustering indicates high viral diversity in terms of gene content.
- No genes are shared between all bins. This suggests there are no core genes.
- some bins share some PCs, and might be closely related
Example heatmap
{kind=link}
Question 3: How can we assign taxonomy to our bins?
Right now we see a few bins that are perhaps related to each other because they share a few proteins. So we know they are similar, but not what they are. Can you think of a way to annotate these sequences through clustering?
Solution
To annotate our contigs, we need to add sequences with known taxonomy and check if they cluster with our bins.
In other words, we need to include our RefSeq protein clusters and genomes.
Would you add all RefSeq genomes? Why (not)?
Hint: check how many RefSeq genomes are in our protein cluster dataset.
Next, we are going to add crAssphage genomes and protein clusters from our RefSeq set, to see if we can find any crAssphage-like phages in our assembled bins.
Question 4: Heatmap with crAssphage RefSeq genomes
follow steps in the R script until the end of the script.
Look at the new heatmap. Interpret what you see.Solution
- We see a couple of different clusters of RefSeq crAssphage Genomes
- Some of our bins cluster with these crAssphages, so perhaps these sequences are also crAssphages?
Example heatmap with crAssphages
{kind=link}
Challenge: Try to classify more bins by including other RefSeq sequences
See if you can annotate any of the other bins by a similar method.
Include PCs and genomes for another viral genus or species that you expect to be present in the human gut microbiome (use google to find which phages you would expect).
Modify the script to filter the RefSeq sequences for that taxon, and make a heatmap. Can you classify any of the other bins?
Key Points