Abundance profiles
Overview
Teaching: 15 min
Exercises: 15 minQuestions
How does the number of reads aligned relate to the abundance of the scaffold?
Objectives
Identify scaffolds present in many samples.
To start off, we will run the python script bam_to_profile.py to get a counts table like the one showed in the previous section. It will print in the terminal the 15 most ubiquitous scaffolds, ie. the scaffolds that are present in more samples. What does the column nsamples mean? Which sample seems to be different?
# create a directory to store output files
$ cd ..
$ mkdir 3_profiles
# run the script
$ python python_scripts/bam_to_profile.py -b 2_mapping/all_samples_cross_sorted.bam -o 3_profiles/
Discussion: Genome fragments
There are several scaffolds present in 11 samples. Could they come from the same organism? In that case, think about reasons for the assembler not being able to reconstruct the complete genome but fragments of it.
The number of reads aligned to a scaffold reflects its abundance (ie. the abundance of the species) in the sample. The more present a species is in a sample, the more sequencing reads is going to obtain during the sequencing process. This abundance is directly related to the depth of coverage.
In the sample below, red species is the most abundant, followed by the green one, and yellow, blue and pink with the same and least abundance. After isolating the DNA and sequencing it, if assemble the reads and map them back to the scaffolds, those representing the red species achieve the highest mean depth (~8x in this example), followed by the green ones (4x) and the rest of species (~2x).
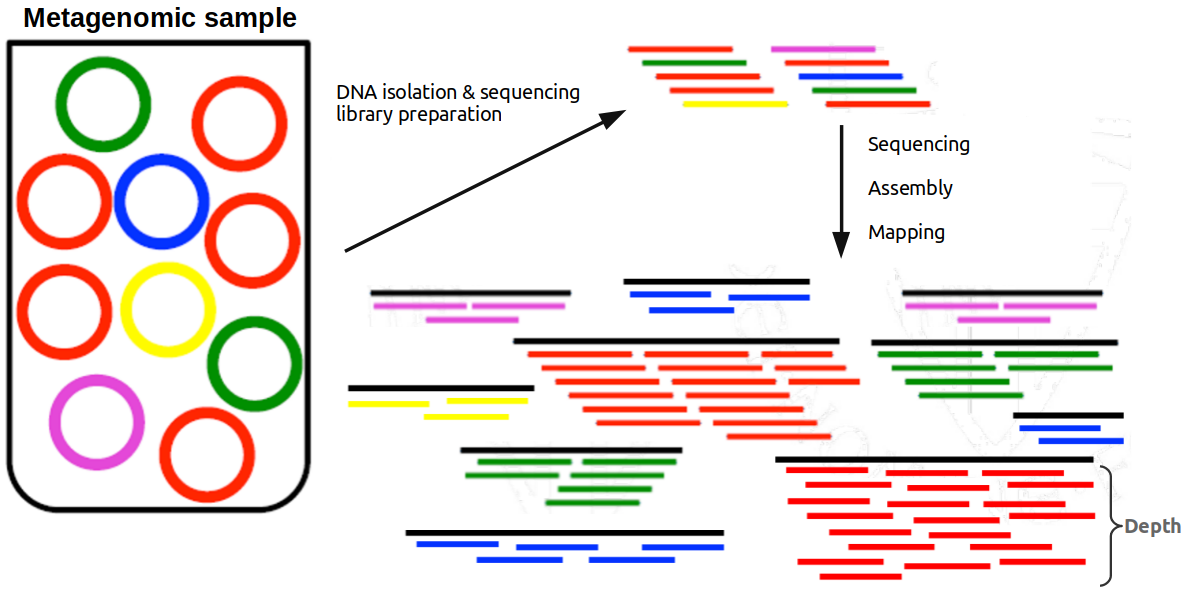
Related to the discussion above, it is interesting to look at the abundance of the scaffolds in a sample because scaffolds coming from the same species should have similar abundances (or depths of coverage, remember they are directly related). It this way, we can group scaffolds in bins representing a genome in a process called binning.
Discussion: Binning
We just saw how abundance can be used to gather scaffolds into a bin to have a more comprehensive representation of a genome. But, what would you do to bin yellow, blue and pink scaffolds? Note well they have the same abundance and will end up in the same bin. Can you think of other features useful for binning?
In the next section we will use the abundances across all the samples to bin our scaffolds.
Key Points
Generally, the higher the number of reads aligned to a scaffold, the higher its abundance in a sample.