Homology based search
Overview
Teaching: 0 min
Exercises: 30 minQuestions
If we classify our viral sequences with Blast, in which cases do you expect that blast sequence searches result in good hits? What would you do if your sequence hit different viral (or bacterial) sequences with low coverage of your query sequence, or low sequence identity with the hit?
Objectives
Run megablast on viral contigs
After you’ve assembled metagenomes (day 1) and identified putative viral contigs (day 2), you probably want to know which viruses you’ve recovered – that is, assign taxonomy. The first thing you might want to do is check whether it is a known virus by searching for highly similar viruses in any of the databases. From day 2 you might remember that homology-based searches will only get you so far for identifying uncultivated viral sequences, as a large part of the virosphere is still uncharted. But if your virus has been sequenced & described before you probably want to know that, because it can save you a lot of time.
Blast a few of the binned contigs (from day 1) with Blast by opening the file in a text editor and copying (part) of the fasta sequence in the browser in the field marked ‘Enter Query Sequence’ (see figure “A”). Another option is to use the terminal and select & copy the sequence from the terminal:
# go to folder containing bins from day 1
cd /path/to/bins/from/day1
# check which files are present
ls
# pick a bin (e.g. bin_03.fasta) and look at the first 150 lines of that file
head -100 bin_03.fasta
# or use a combination of 'head' and 'tail' to look at a part somewhere in the middle:
head -4000 bin_03.fasta | tail -250
# Note:
# bins typically contains multiple contigs. If you want to blast multiple contigs,
# just make sure that they contain the header lines (starts with ">") in between.
# You can select the results for each of the sequences on the blast results page.
# Note:
# Don't do this for many or very long sequences since we don't want to overload the blast server.
# If you want to search a lot of sequences in this way (NOT recommended!) you would
# use a command line blast version on your own server.
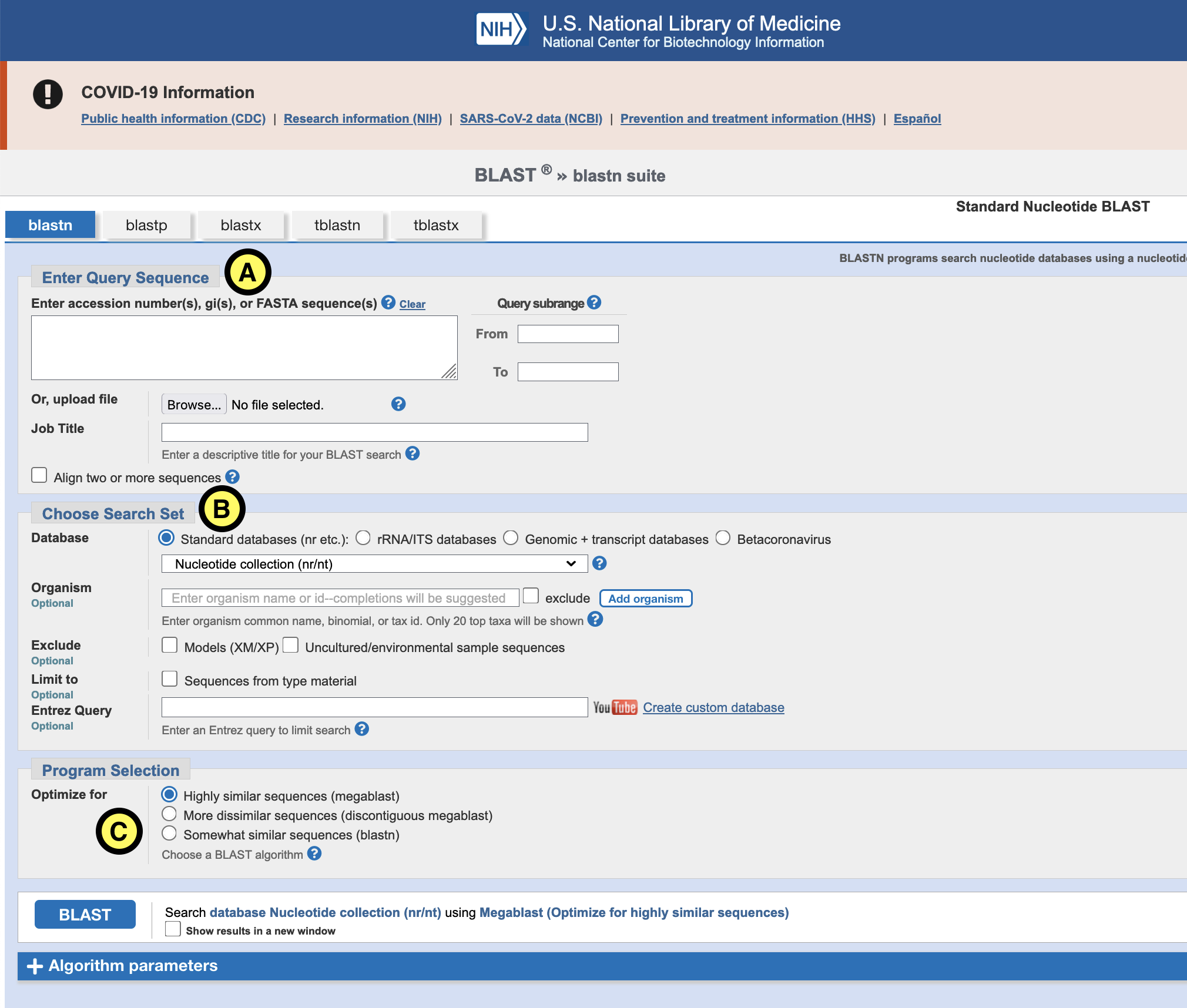
For “Database” (B) select Nucleotide collection (nr/nt) or RefSeq Genome database. For ‘Programs selection’ (C) choose Megablast. This is the least sensitive, but also the fastest of the different blast flavors.
Click the blue “Blast” button and wait for the results.
Question: Blast results
Inspect the “Descriptions”, “Graphic summary” and “Taxonomy” tabs to answer the following questions:
- What is the top hit? Do you have a single good hit or multiple hits?
- What is the length of aligned region in the query and target?
- What is the percent identity?
- Can you assign the contig to a taxonomic group? Why (not)?
Try a few different sequence parts of the bin, and try a few other bins. Discuss what you find with your neighbour.
Key Points
Sometimes you might find a good hit, for example for the crAssphage bins, or for many of the well-described viruses infecting humans. In other cases, we need more sophisticated search strategies to assign a given viral sequence to a previously described taxon.