Assembly lecture
Overview
Teaching: 60 min
Exercises: 30 minObjectives
Watch the lecture videos and read about assembly algorithms
![]()
Sequence assembly is the reconstruction of long contiguous sequences (called contigs or scaffolds, see video below) from short sequencing reads. Before 2014, a common approach in metagenomics was to compare the short sequencing reads to the genomes of known organisms in the database (and some studies today still take this approach). However, this only works if the organisms in the database are closely related to the ones in the metagenomic sample. Recall that most of the sequences in a metavirome are unknown (“viral dark matter”), meaning that they yield no matches when compared to the reference database. Because of this, we need database-independent approaches to reconstruct new viral sequences. As sequencing technology and bioinformatic tools improved, sequence assembly enabled the recovery of longer sequences from metagenomic data. Having a longer sequence means having more information to classify it, so using metagenome assembly helps to characterize complex communities.
Video on sequence assembly
Watch the lecture video “Assembly strategies for genomics and metagenomics”. It will introduce reference-guided and de-novo assembly of genomic and metagenomic sequences (56 minutes):
Discussion
Watch the lecture video below and write down at least 3 questions and/or discussion points about it.
- Click on the image to see lecture video “Assembly strategies for genomics and metagenomics” by Prof Bas E. Dutilh (56 minutes):
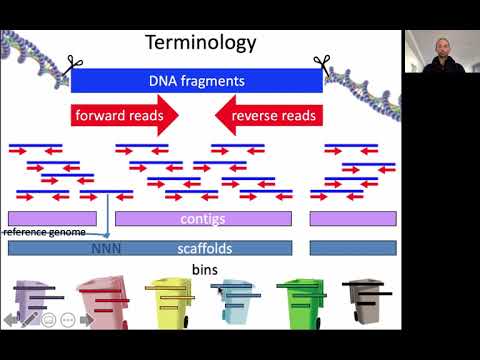
Questions
- Would you use DBG (De-Bruijn Graph) or OLC (Overlap-Layout-Consensus) to assemble a dataset consisting of one billion short sequencing reads?
- What are the strengths and weaknesses of reference-guided assembly and de novo assembly?
- Would you use reference-guided or de novo assembly to assemble the genome of a model organism to discover mutations that occurred during an evolutionary experiment?
- Would you use reference-guided or de novo assembly to determine the genome sequence of an unknown organism?
- Why does metagenome assembly generally yield shorter contigs than genome assembly?
Additional reading: Computational Biology: Genomes, Networks, Evolution MIT course 6.047/6.878 (Prof. Manolis Kellis). This book is part of a course on Computational Biology and contains several topics that are relevant for Bioinformatics.
Read and summarize
Read the following sections and summarize shortly (less than half a page per section) their key points in your lab book:
- “5.2 Genome Assembly I: Overlap-Layout-Consensus Approach” and “5.3 Genome Assembly II: String graph methods” (pages 93 to 102).
Key Points
Sequence assembly can be used to assemble genomes from reads
Metagenome assembly generally yields shorter contigs than genome assembly