Gene Calling and Functional Annotation II
Overview
Teaching: min
Exercises: 180 minObjectives
Perform gene calling and functional annotation on the viral contigs
Running pharokka
For the functional annotation of our viral contigs, we will be using Pharokka. The “viral contigs” are the contigs we filtered yesterday (i.e. using Jaeger and CheckV)
We first have to load the appropriate conda environment
source /vast/groups/VEO/tools/anaconda3/etc/profile.d/conda.sh
conda activate pharokka_v1.7.1
The base usage for this program follows:
pharokka.py -i <fasta file> -o <output folder> -d <path/to/database_dir> -t <threads>
See the docs for the pharokka parameters
We have to add the following options:
- the pharokka database is here:
/work/groups/VEO/databases/pharokka/v1.7.1/ - To run on viral contigs from a metavirome:
-m - To also use PyHMMER
--meta_hmm - To use Phanotate for the gene prediction:
-g phanotate
Therefore your command should look like this:
pharokka.py -i filtered_assembly.fasta -o output_dir -d path/to/database -t 10 -m --meta_hmm -f -g phanotate
We recommend modifying the following SBATCH resources:
- #SBATCH –cpus-per-task=10
- #SBATCH –partition=short,standard,interactive
- #SBATCH –mem=10G
Exercise
- Run pharokka with the appropriate parameters using an sbatch script
sbatch script for pharokka
#!/bin/bash #SBATCH --tasks=1 #SBATCH --cpus-per-task=10 #SBATCH --partition=short,standard,interactive #SBATCH --mem=20G #SBATCH --time=02:00:00 #SBATCH --job-name=pharokka #SBATCH --output=./1.4_annotation/10_pharokka/pharokka.slurm.out.%j #SBATCH --error=./1.4_annotation/10_pharokka/pharokka.slurm.err.%j source /vast/groups/VEO/tools/anaconda3/etc/profile.d/conda.sh && conda activate pharokka_v1.7.1 database='/work/groups/VEO/databases/pharokka/v1.7.1/' assembly='./1.3_virus_identification/40_results_filter_contigs/assembly.fasta' outdir='./1.4_annotation/10_pharokka' # extra options to use # -m: run in metavirome mode (each contig is a new virus) # --meta_hmm : also run PyHMMER on the PHROGS (richer annotations with this) # -f: force restart the outputs # -g: gene annotation by phanotate (default for meta mode is prodigal-gv) pharokka.py -i $assembly -o $outdir -d $database -t 10 -m --meta_hmm -f -g phanotate
Plotting functional annotations
Choose a pet contig (or two)
Exercise - extracting contig information
- Choose a pet contig. Maybe your favourite contig number… but we recommend choosing one with high completeness and which is ‘annotation-rich’
- perhaps with many annotated genes, a crispr region, mash results (from pharokka) etc.
- Discuss with your classmates so that everybody picks a different virus one. We will continue using this “pet” contig in the future lessons also. a) Write down some information about your contig. e.g. contig length, # of genes, # of annotated genes, gc%, coding density
For the plotting we, we will still use pharokka. Pharokka’s built-in pharokka_plotter.py uses a python library called pyCirclize. You will also use this library later for the visualization homework so stay tuned!
pharokka_plotter.py requires a gff, genbank and fasta file for the viral contig. Therefore, we have to extract these from the larger pharokka.gff and pharokka.gbk outputs.
The .gff file contains information about the gene positions and strands
The .gbk file contains information about the functional annotations of each gene
The fasta file contains the nucleotide sequence
Exercise - extracting contig information
We need 3 pieces of information about your particular contig to use the pharokka plotter
- contig.gff : extracted from the pharokka.gff
- contig.gbk : extracted from the pharokka.gbk
- contig.fasta : nucl. sequence extracted from the filtered assembly
- Extract the functional annotation information for your contig from the
pharokka.gffandpharokka.gbkfiles and the contig nucleotide sequence from the fasta file
Gff and fasta files are easy to pull information from so we can just grep or sed the contig directly into an output file
Extract contig.gff
grep "contig_xxxx" pharokka.gff > contig_xxxx.gff # for example mkdir -p ./1.4_annotation/10_pharokka/11_selected_contig # get the contig.gff grep "contig_827" ./1.4_annotation/10_pharokka/pharokka.gff > ./1.4_annotation/10_pharokka/11_selected_contig/contig_827.gff
Extract contig fasta
# if your fasta file is in multi-line format # this tells sed to print between "contig_1026" and the next ">", including both patterns # `head` removes the last line (the second header) sed -n '/contig_1026/,/>/p' phage_contigs.fasta | head -n -1 # for example: sed -n '/contig_827/,/>/p' ./1.2_assembly/10_results_assembly_flye/cross_assembly/assembly.fasta | head -n -1 > ./1.4_annotation/10_pharokka/11_selected_contig/contig_827.fasta
Genbank files are a bit more difficult to extract information from but they are formatted sequentially by records. Luckily for us, there are pre-built modules in the biopython library to work with genbank files. SeqRecord and Bio.SeqIO.parse
If you head the pharokka.gbk file, you might see how the records are structured:
LOCUS contig_1026 35733 bp DNA linear PHG 06-SEP-2024
DEFINITION contig_1026.
ACCESSION contig_1026
VERSION contig_1026
KEYWORDS .
SOURCE .
ORGANISM .
.
FEATURES Location/Qualifiers
CDS complement(618..749)
/ID="contig_1026_CDS_0001"
/transl_table=11
/phrog="No_PHROG"
/top_hit="No_PHROG"
/locus_tag="contig_1026_CDS_0001"
/function="unknown function"
/product="hypothetical protein"
/source="Pyrodigal-gv_0.3.1"
/score="3.3"
/phase="0"
/translation="MTRWNTTLAVYEIWTGTQWQAVASSTYSINYLIVAGGASGAHY*"
See if you can write a script to extract the genbank record for your contig using this module - for clues - see solution below!
The whole contig is called a “record” - see the biopython SeqRecord class page for what each element in the record is called, use Bio.SeqIO.parse to iterate through these records and Bio.SeqIO.write to write out the record you want.
biopython script to extract contig.gbk (genbank)
#!/usr/bin/python3 from Bio import SeqIO import sys # read files in as variables input_gbk = sys.argv[1] output_gbk = sys.argv[2] contig = sys.argv[3] # parse the genbank and write output for record in SeqIO.parse(input_gbk, "genbank"): # print(record.id) if record.id == contig: SeqIO.write(record, output_gbk, "genbank")
sbatch script to run your python script
#!/bin/bash #SBATCH --tasks=1 #SBATCH --cpus-per-task=1 #SBATCH --partition=short,standard,interactive #SBATCH --mem=2G #SBATCH --time=00:01:00 #SBATCH --job-name=py_script #SBATCH --output=./1.4_annotation/10_pharokka/extract_contigs.slurm.out.%j #SBATCH --error=./1.4_annotation/10_pharokka/extract_contigs.slurm.err.%j # activate the python virtual environment with the packages we need source ./py3env/bin/activate # change the output and contig names accordingly python3 python_scripts/annotation/extract_contig_gbk.py pharokka.gbk output_contig.gbk contig_xxxx
Plot your contigs!
Once we have our fasta, gff and genbank files, we can use pharokka_plotter.py to plot our pet contig. pharokka_plotter.py requires the same conda environment as pharokka. So, to to activate the environment use the same commands you used for pharokka. Feel free to experiment with the different options given in the manual. The base usage is:
pharokka_plotter.py -i input.fasta -n pharokka_plot --gff pharokka.gff --genbank pharokka.gbk
You no longer need the pharokka database and we recommend modifying the following SBATCH parameters #SBATCH --mem=10G
Exercise - plotting
- Plot the genome annotations for your pet contig
Make sure to add your plot into your lab book!
sbatch script for all the extraction steps + pharokka_plotter
#!/bin/bash #SBATCH --tasks=1 #SBATCH --cpus-per-task=10 #SBATCH --partition=short,standard,interactive #SBATCH --mem=10G #SBATCH --time=2:00:00 #SBATCH --job-name=pharokka_1 #SBATCH --output=./1.4_annotation/10_pharokka/pharokka_plotter.slurm.out.%j #SBATCH --error=./1.4_annotation/10_pharokka/pharokka_plotter.slurm.err.%j assembly='./1.2_assembly/10_results_assembly_flye/cross_assembly/assembly.fasta' outdir='./1.4_annotation/10_pharokka/11_selected_contig' mkdir -p $outdir # get the contig.gff and contig.fasta grep "contig_827" ./1.4_annotation/10_pharokka/pharokka.gff > $outdir/contig_827.gff sed -n '/contig_827/,/>/p' $assembly | head -n -1 > $outdir/contig_827.fasta # next, we get the contig.gbk using a python script # change the names accordingly # activate the python virtual environment with the packages we need source ./py3env/bin/activate # the inputs for the following python script should be # python3 script.py input.gbk output.gbk contig_name input_gbk='./1.4_annotation/10_pharokka/pharokka.gbk' output_gbk='./1.4_annotation/10_pharokka/11_selected_contig/contig_827.gbk' name='contig_827' python3 python_scripts/annotation/extract_contig_gbk.py $input_gbk $output_gbk $name # Then we run pharokka plotter on our selected contig source /vast/groups/VEO/tools/anaconda3/etc/profile.d/conda.sh && conda activate pharokka_v1.7.1 # define all your file paths for readability outdir='./1.4_annotation/10_pharokka/11_selected_contig/' fasta='./1.4_annotation/10_pharokka/11_selected_contig/contig_827.fasta' gff='./1.4_annotation/10_pharokka/11_selected_contig/contig_827.gff' gbk='./1.4_annotation/10_pharokka/11_selected_contig/contig_827.gbk' pharokka_plotter.py -i $fasta -n $outdir/pharokka_plot --gff $gff --genbank $gbk
The final plot might look something like this:
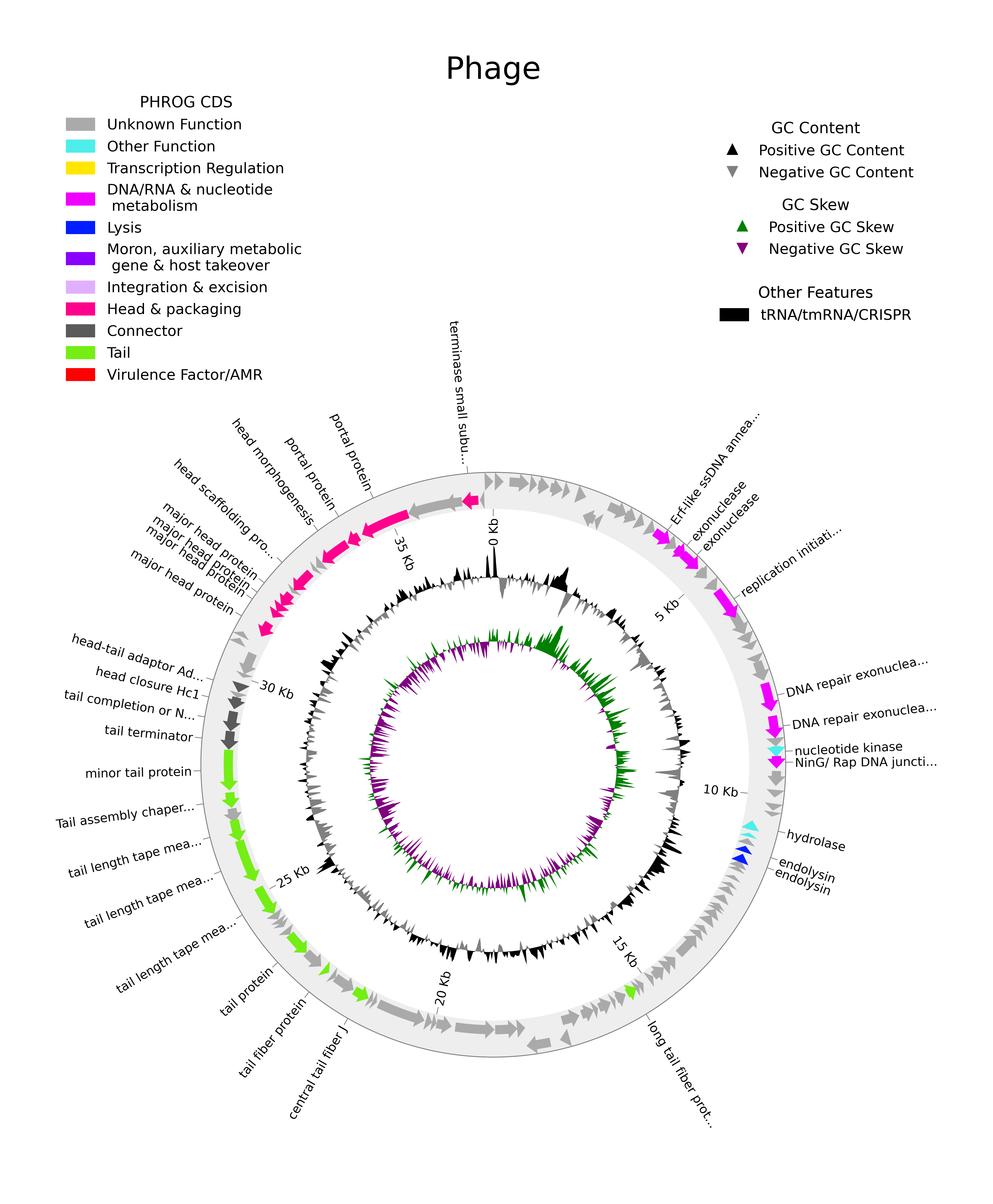
Interpreting your contig
With functional annotation we can begin to get an insight into our viruses to piece together information about their lifestyles, hosts interactions etc, what type of genes they have. You’ll find that most genes on viral contigs are not actually annotated - this is because 1) viruses evolve very quickly and assigning gene annotations by homology is difficult, and 2) viruses are highly mosaic - they pick up and lose genes from their hosts and other viruses, meaning not all viruses have all the same genes and viral gene databases do not always account for their variability. The creation of newer tools like phold aid in this by using structural homologs to assign gene annotation.
Note : We have phold installed on Draco, if you would like to experiment with it for your project! For example, you might pick a contig that is NOT well annotated and see how many extra genes you can annotate using structural homology
Exercise - Interpret your contigs
- How many structural genes are annotated?
- Describe at least 2 different types of structural genes a) Where are they placed in your genome? b) To which part of phage structure do they belong? (capsid, tail etc) c) What other viruses have these structural features?
- How many replication genes are annotated?
- These could be genes related to transcriptional regulation, DNA/RNA metabolism lysis etc
- Describe the role of 1 replication gene a) What is the function of the gene? b) What part of the replication cycle is it used? c) How does it interact with the host genes (if they do)
- Describe 1 or more interesting genomic features
- CRISPR arrays, tRNA, virulence factors etc
- To the best of your ability, infer how this virus might be interacting with its host.
- You might investigate clues temperate/lytic lifestyles, auxiliary metabolic genes, anti-crisprs etc
Key Points
Functional annotation of viral genomes can give clues about viral lifestyle and host interactions